DrugLLM——利用大规模语言模型通过 Few-Shot 生成生物制药小分子
摘要
小分子由于能够与特定的生物靶点结合并调节其功能,因此在药物发现领域发挥着至关重要的作用。根据美国食品和药物管理局(FDA)过去十年的审批记录,小分子药物占所有获批上市药物的 76%。小分子药物的特点是合成相对容易,生物利用度高,易于到达预定靶点。然而,设计具有理想特性的分子非常困难,需要耗费大量的资源和时间。例如,找到一种有效的药物需要 9-12 年的药物开发过程和数十亿美元。
寻找新分子的范围非常广泛,可合成的药物分子多达 1060 种。因此,对于化学家来说,确定与生物靶标相互作用的分子是一项重大挑战。现代技术可以在实验室中测试106 种以上的分子,但更大规模的实验却过于昂贵和不切实际。因此,需要使用计算工具来缩小搜索范围。
虚拟筛选就是这样一种工具,它可以从数百万个现有分子和虚拟分子中找出有潜力的分子。然而,高通量筛选和虚拟筛选只能针对已知的、可合成的分子,无法产生新的分子。
作为探索这一庞大候选分子库的替代方法,从头设计展示了生成完全新颖独特分子的能力。传统的从头设计根据受体和配体结构生成新分子,但最近深度学习和强化学习已显示出前景。特别是,集成生成和预测神经网络等方法正被用于生成新分子。
尽管这些新技术取得了进展,但通过富彻特生成分子的方法仍未得到充分研究。利用富彻特法生成分子的目的是从有限的分子样本中生成具有预期特性的新分子。
目前的许多从头设计都需要成千上万的数据进行训练,而在药物发现领域,数据通常很少。因此,能够在四次扫描中进行生成对于从头设计技术的发展至关重要。
大规模语言模型在自然语言处理领域取得了长足进步,尤其是在福克索特学习问题上,但在处理生物和化学语言时仍面临挑战。因此,本文提出了用于药物发现的大规模语言模型 DrugLLM。
DrugLLM 使用基于组的分子表示法(GMR)来表示分子并解决 SMILES 中固有的问题 GMR 使用结构组来构建分子的拓扑结构,并将其转换为线性序列。GMR 还可用于创建根据特定分子特性进行修饰的分子序列。它还能根据特定的分子特性组织修饰序列。DrugLLM 可以根据下一个分子的修改历史不断预测其修改顺序,从而学习分子结构与性质之间的关系。 DrugLLM 是第一个通过 Fewshot 生成分子的大规模语言模型。
数据收集和准备
为了训练和分析 DrugLLM,我们使用 "ZINC "和 "ChEMBL "数据集建立了大型数据集;ZINC 是一个免费数据库,其中包含 230 多万种化合物,可以购买三维格式进行对接。从该数据库中筛选出药物样分子(具有药物常见理化性质和结构特征的分子),得到 450 万个分子。生物活性数据是按照 Stanley 等人(2021 年)的预处理方法收集的,排除了非药物类化合物,并应用了标准的清洗和规范化处理。此外,所有分子都用 SMILES 字符串表示,并标注特定属性。
为了便于进行特性比较,只考虑以实际值表示的特性类别。这就产生了一个由数千个表格数据组成的大型数据集,每个表格包含数百个具有相同特性的被测分子。根据收集到的数据,这些数据被转换成有意义的文本句子和段落。
例如,结构相似的两个分子之间的修改被视为一个句子,而多个修改案例被视为一个段落。同一段落中的修改被假定为描述了相同的性质变化。如果前两个修改情况描述的是溶解度的增加,则该段中的所有其他句子也被假定为溶解度的增 加。
这一规定是通过启发式算法实现的。首先,根据一组具有某种特征的分子,使用随机选择的聚类中心对分子进行聚类。如果某个分子与某个中心的相似度超过 0.6,该分子就会被聚类到该中心。聚类中心的数量会动态增加,直到该集合中的所有分子都被分类为止。
此外,除了对单一属性进行分子修改外,还考虑了多种属性的组合。这些主要涉及可通过 Python 脚本计算的简单分子特性。例如,LogP、拓扑极性表面积(TPSA)及其组合都包含在训练集中。为建立训练数据集,总共收集了超过 2500 万个修改段落和 20 亿个分子。该数据集包含 1 万多种不同的分子性质、活性和组成;除了 SMILES 分子外,每个段落还包含对性质优化的附加说明,以便将性质的含义与分子结构联系起来。
基于组的分子表征(GMR)
基于组的分子表征(GMR)框架用于分子表征。其目的是通过将分子分解为结构组并记录其连接信息,从而在组的基础上重建SMILES 字符串,从而提高分子信息的可解释性。

首先,利用 ChEMBL 数据库收集分子数据。然后,使用 SMILES 表示法提取分子内的环状结构信息,并对相交的环进行整合,以识别特定的结构组。对于非环部分,所有的 C-C 键都会被裂解,剩余的分子片段被视为独立的结构组。这样就可以创建一个综合字典,为每个组分配一个唯一的字符串标识符。
然后将单个分子的 SMILES 字符串拆分成若干结构单元。使用宽度优先搜索算法,检查分子在去除结构单元后是否仍然相连,并记录连接点上的两个原子。这就形成了原子的特征。对每个结构组和拆分后的分子片段进行 SMILES 归一化处理,并将字典中的相应字符串合并为一个编码字符串。如此反复,最终生成准确的分子编码�
根据编码的分子片段,参照记录的拼接信息,在正确的位置上重组每个结构组。这一过程不断重复,直到所有结构组都被正确拼接,最后对原始分子 SMILES 进行解码。这样做的目的是确保分子信息的完整性和可逆性。

这有效地管理了分子的详细结构信息,提高了分析的准确性。
实验和结果
本文的重点是学习能够捕捉分子结构与相应化学和生物活性之间关系的大规模语言模型:ChatGPT 和 LLaMA 是使用互联网上的大量文本数据进行训练的,DruggGPT 使用 SMILES 作为分子表征,而 DrugLLM 则使用基于组的分子表征(GMR)作为其主要语言表征。而 DrugLLM 则使用基于组的分子表征 (GMR) 作为其主要语言表征。这种GMR 通过使用结构组来表示分子结构,克服了 SMILES 符号的三大难题。
首先是令牌数量庞大:在 SMILES 格式中,每个字母都被视为一个单独的令牌,因此令牌数量庞大,在训练过程中会消耗大量计算资源;其次是环结构复杂。分子内环结构的表示尤其复杂,使得模型难以训练。即使分子结构发生微小变化,也会导致相应的 SMILES 表示法出现巨大差异。
如下图所示(转载),GMR 框架使用独特的字符串标识符来表示不同的结构组,这些结构组通过斜线环绕的数值位置数据联系起来。标记,从而减少标记的数量。GMR 还可以通过整合和去除循环结构来简化分子组装逻辑,从而降低模型识别的难度。此外,SMILES 字符串中的细微结构变化所造成的差异也可以最小化。
为了训练 DrugLLM,我们构建了由分子修饰组成的句子和段落作为训练数据,如下图所示。具体来说,DrugLLM 将结构相似的两个分子之间的修饰视为一个句子,将一系列此类修饰视为一个段落。段落中的分子修饰必须描述相同的性质,例如,如果前三个分子修饰样本描述了氢键受体数量的增加,那么预计该段落中的后续句子也将描述受体数量的增加。这样,段落的内容就会集中起来,DrugLLM 就能根据之前的上下文自回归地预测下一个标记。此外,由于每个段落都包含各种分子特性,而且每个段落都涉及自身的分子特性,DrugLLM 需要具备在上下文中学习的能力。

然而,相关的数据集却很少见。本文从 ZINC 数据库和 ChEMBL 平台收集了表格形式的分子数据集,并将其转换成相应的句子和段落。总共收集了超过 25 000 000 000 个修改过的段落和 200 000 000 000 个分子作为训练数据集。

该数据集包含 10,000 多种不同的分子特性和活性,包括氢键受体数量和拓扑极性表面积 (TPSA)。基于对最先进的大规模语言模型的预训练,DrugLLM 采用了 Transformer 架构。它还采用了 LLama 7B 参数,并通过引入常用的 SMILES 标记来扩展词汇量。使用 AdamW 优化器,DrugLLM 在八台英伟达 RTX 3090 GPU 上进行了为期六周的训练。从机器学习的角度来看,该段落充当了 Fewshot 的分子生成过程。因此,经过训练的 DrugLLM 可以通过 Fuchots 生成分子,而无需进一步微调。
DrugLLM是一个利用"快速学习"(Few-Shotlearning)优化理化性质的模型。如下图所示,K-Shot 学习为模型提供了 K 对改良示例和基准分子。该模型的目标是在修改样本的基础上生成具有改进特性的新分子,同时保持与基准分子的结构相似性。由于输入标记的限制,优化示例分子的数量最多不超过 9 对。

为了直观显示生成分子和基准分子在结构上的相似性,我们使用 UMAP(Uniform Manifold Approximation and Projection)方法制作了一张图表。生成分子(左侧)和原始分子(右侧)的分布相吻合,这种分布的相似性和生成分子 LogP 特性的明显改善表明了模型的高性能。

为了评估DrugLLM 的Fewshot生成分子的能力,我们选择了四种理化性质作为测试任务,包括 LogP(水-辛醇分配系数)、溶解度、合成可及性和拓扑极性表面积(TPSA)。这些属性可以通过基于机器学习的脚本进行精确估算,因此被广泛用于评估分子生成模型。
为了进行比较,使用了结点树变异自动编码器(JTVAE)、变异结点树神经网络(VJTNN)和基于支架的分子生成器(MoLeR)。它还包括基于 JTVAE 潜在空间的随机生成控制。生成分子的质量根据成功率和分子的相似性进行评估。成功率代表生成的分子中遵循修改样本规则的比例。为避免生成偏差,输入上下文(语言模型提示)描述了均衡的增减特征。
下图显示了原始数据和生成数据的一些关键属性(LogP、溶解度、合成可达性和 TPSA)的分布情况。这些分布使用核密度估计 (KDE) 进行了可视化。这进一步证明了模型的有效性。

如下图所示,我们还报告了少次生成的性能与 LogP 值的关系:JTVAE、VJTNN 和 MoLeR 这三种基线分子生成模型的成功率约为 50%,与随机生成类似。然而,DrugLLM 在分子生成方面表现出逐步提高的趋势,随着生成次数的增加,生成分子的准确率提高到 75%。分子溶解度、合成可及性和 TPSA 的性能比较也同样一致。

虽然通常很难优化修改较少(相似度高)的分子,但随着生成相似度的增加,DrugLLM 仍然保持了较高的成功率,这表明它在融合生成方面表现出色。此外,DrugLLM-GMR 还略胜于 DrugLLM-SMILES,证明了 GMR 在训练大型模型方面的优势。
此外,如上所述,DrugLLM已显示出通过融合产生具有出色理化特性的分子的能力,这反过来又验证了 DrugLLM 在分子生物活性方面的有效性。与理化性质相比,生物活性被认为是一项更加复杂和具有挑战性的任务,因为 DrugLLM 生成的分子通常都是新的,而且没有记录在 ChEMBL 数据库中。与理化性质不同,生物活性更难通过化学或物理规则来估算。此外,实验室实验所需的大量时间和费用也使大规模分子评估变得困难。因此,本文利用消息传递来预测生物活性。
在使用 ChEMBL 数据库建立药物LLM 数据集之前,先对所有生物活性进行了扫描,并选择了那些样本数量相对充足(N ≥ 800)且属性预测准确(皮尔逊相关系数 r ≥ 0.75)的生物活性。最终选出 10 项活动,并将其排除在训练数据之外。预测模型的皮尔逊相关系数超过 0.75,因此在统计上与实际评级有很好的相关性。
如下表所示,与随机生成相比,三种生成基线并没有实现有意义的改进。这表明这些分子生成模型无法成功捕捉到基于有限样本的修改规则。
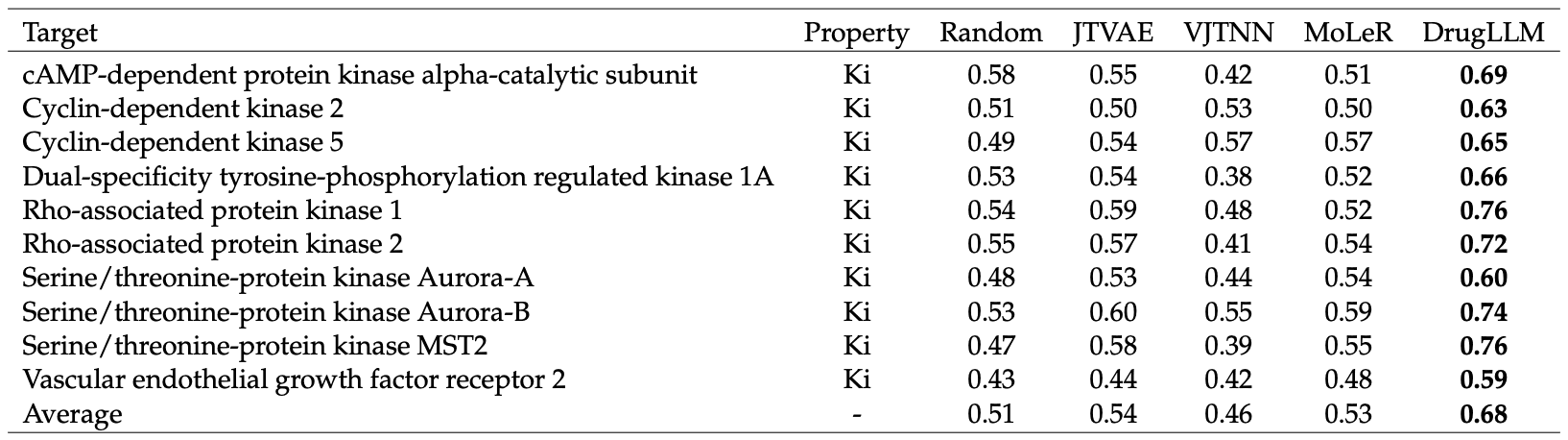
相比之下,DrugLLM 在大多数测试特性上的表现明显优于其他基线。尤其是,DrugLLM 能够以 76% 的成功率生成与 Rho- 相关蛋白激酶 1 结合的适当分子。这些测试特征在 DrugLLM 的训练过程中都没有观察到。这些结果表明,DrugLLM 有能力从数量有限的示例中为未知分子特性找到固有的分子修饰规则。
总结
在本文中,我们讨论了一项新颖的计算任务:通过福柯索进行分子优化。这项任务以感兴趣的分子为基础,从少量修改过的样本中生成新分子。目前已经提出了多种富彻学习任务,但通过富彻生成分子的工作还很少。通过 Fuchots 进行分子优化要求模型能够从少量样本中学习抽象规则,并将其应用于新分子。就目前的方法而言,ChatGPT 和其他分子生成模型并不能很好地完成这项任务,但本文提出的 "DrugLLM "却表现出了卓越的性能。
DrugLLM 是基于大量小分子和生物数据建立的大规模语言模型。最近的大规模语言模型 ChatGPTmAlpaca 和 ChatGLM 在一般自然语言生成方面具有出色的能力,但缺乏生物学和药理学知识。此外,还有一些专门针对生物学和医学的大规模语言模型,但这些模型仍然采用传统的学习策略,没有解决如何理解生物学和化学语言以及如何进行福氏学习的难题。在本文中,DrugLLM 利用 GMR提出了一种迭代的、对上下文敏感的分子修饰新方法。
不过,这种方法也有一些局限性。首先,由于硬件限制,DrugLLM 最多只能支持九次分子修饰。此外,DrugLLM 的零次分子优化仍处于早期阶段,有待改进。目前,DrugLLM 只能根据两个已知的分子特性来优化分子。此外,目前使用的 GMR 难以表现特定情况下的复杂分子,也缺乏标准化方法。
作者指出,"DrugLLM"是 Fewshot 首次推出的用于分子生成和优化的大规模语言模型。作者从与分子特性和生物活性相关的数据中构建了一个大型文本语料库,以自回归方式训练 DrugLLM;DrugLLM 的出色表现表明,它作为药物分子发现领域的强大计算工具具有巨大潜力。
注:
论文地址:https://arxiv.org/pdf/2405.06690
原文地址:https://ai-scholar.tech/en/articles/large-language-models/drugllm
原文地址:https://blog.csdn.net/matt45m/article/details/143893076
免责声明:本站文章内容转载自网络资源,如本站内容侵犯了原著者的合法权益,可联系本站删除。更多内容请关注自学内容网(zxcms.com)!