单细胞copyKat分析学习和整理
CopyKAT(肿瘤拷贝数核型分析)是一种使用综合贝叶斯方法的计算工具,能够在单细胞中以5MB分辨率检测全基因组非整倍体,以便从高通量单细胞RNA测序数据中区分肿瘤细胞与正常细胞,并识别肿瘤亚克隆。
(这里提一下,“5MB”是指 5兆碱基对(5 megabase pairs, 5 Mb),表示基因组上5百万个碱基对的区域。这个分辨率意味着CopyKAT可以在单细胞中检测到跨越5百万碱基对的染色体拷贝数变化,进而识别该区域内的非整倍体(基因组拷贝数异常)。在基因组分析中,分辨率越高(即Mb数越小),就越能检测到更小范围的拷贝数变化。5Mb分辨率是一种适中的水平,能够提供足够的精度来区分肿瘤细胞和正常细胞,并检测肿瘤亚克隆的拷贝数变异)。
其推断RNAseq数据中的DNA拷贝数事件的基本逻辑是,通过相邻基因的表达水平提供深度信息,从而推断该区域的基因组拷贝数。CopyKAT估算的拷贝数谱与全基因组DNA测序获得的实际DNA拷贝数可以达到80%的高度一致性。区分肿瘤/正常细胞状态的原理在于非整倍体在90%的癌症中普遍存在。具有广泛全基因组拷贝数异常(非整倍体)的细胞被视为肿瘤细胞,而基质正常细胞和免疫细胞通常具有2N二倍体或近二倍体的拷贝数谱。

那么这个工具跟目前另一个常用的工具inferCNV之间有什么区别呢?
inferCNV是使用邻近基因表达水平的变异来推断细胞中的CNV,基于假设拷贝数变化会引起局部基因表达的显著变化。inferCNV不使用贝叶斯方法,而是基于基因表达值与正常参考细胞进行比较,识别异常表达模式来推断拷贝数变化。它需要预先指定一组正常细胞作为参考,这些参考细胞用于构建基线表达模式,再通过与肿瘤细胞的表达模式比较来推断CNV。因此,inferCNV在依赖参考细胞的选择上比CopyKAT更为严格。
在应用层面两种都可以使用~ 不必纠结。
分析步骤
1.导入
rm(list = ls())
library(copykat)
library(Seurat)
library(gplots)
library(ggplot2)
library(qs)
#这里加载的是seurat对象,替换自己的数据即可
#load("scRNA.Rdata")
scRNA <- qread("sc_dataset.qs")
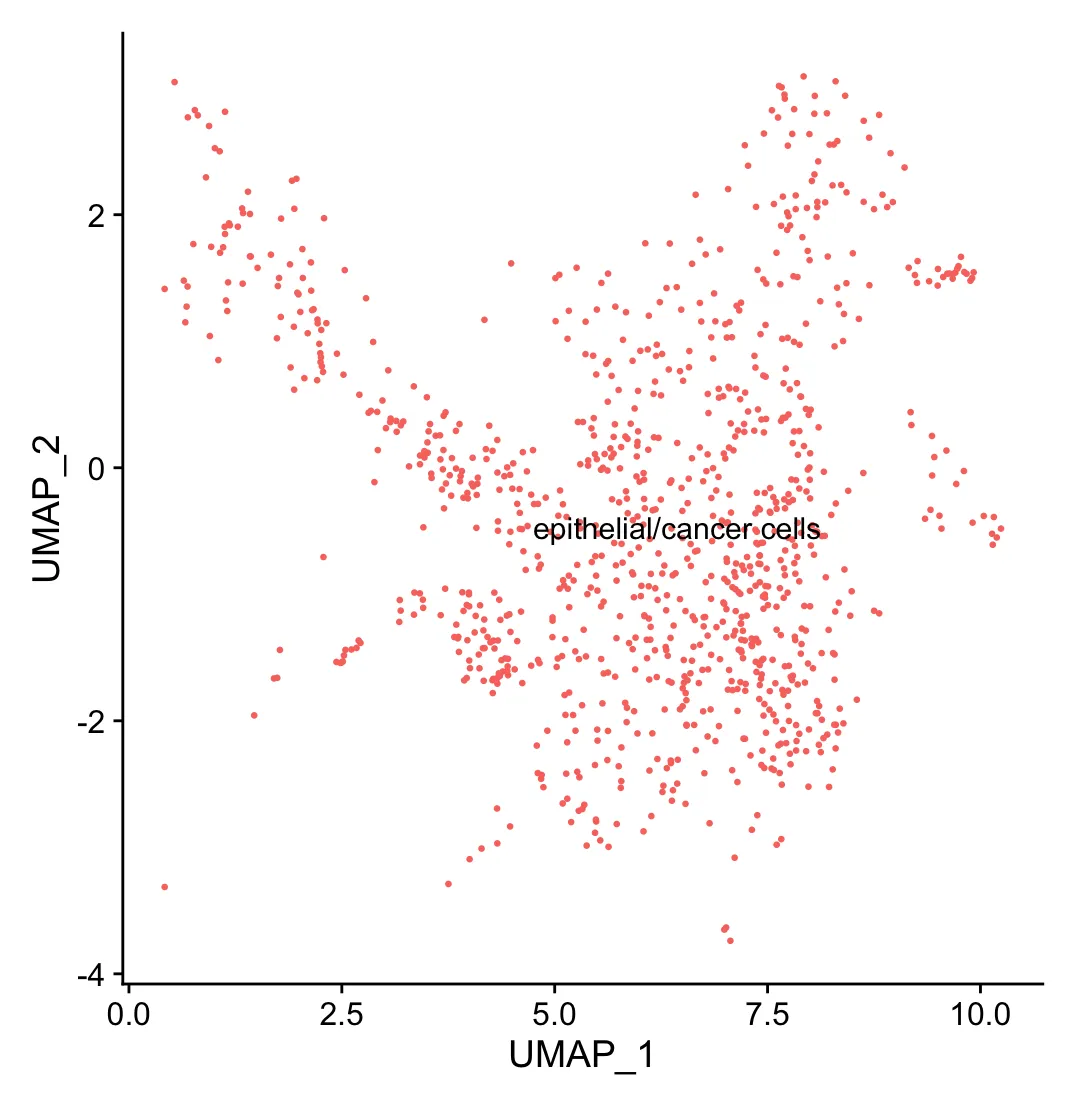
# 提取肿瘤细胞并抽样一下
scRNA <- subset(scRNA,celltype=="epithelial/cancer cells")
scRNA <- subset(scRNA,downsample = 1000)
#检查一下自己导入进来的数据
DimPlot(scRNA,reduction = 'umap',
label = TRUE,pt.size = 0.5) +NoLegend()
2.数据预处理
exp.rawdata <- as.matrix(scRNA@assays$RNA@counts)
3.运行CopyKat
这一步很慢
copykat.test <- copykat(rawmat=exp.rawdata,
id.type="S", # S是symbol的含义
ngene.chr=5, # 规定每条染色体中至少5个基因来计算DNA拷贝数(可调整)
win.size=25, # 每个片段至少25个基因
KS.cut=0.1, # 增加KS.cut会降低敏感度,通常范围在0.05-0.15
sam.name="241016", # 自行命名
distance="euclidean",
norm.cell.names="",
output.seg="FLASE",
plot.genes="TRUE",
genome="hg20",
n.cores=8)
4.提取结果
二倍体通常指正常的非癌细胞,而非整倍体则指具有染色体数量异常、可能属于癌细胞的细胞
pred.test <- data.frame(copykat.test$prediction)
# check一下数据
table(pred.test$copykat.pred)
# aneuploid diploid not.defined
# 348 450 202
# 提取aneuploid(非整倍体)/diploid(二倍体)细胞
pred.test <- pred.test[which(pred.test$copykat.pred %in% c("aneuploid","diploid")),]
CNA.test <- data.frame(copykat.test$CNAmat)
查看pred.test$copykat.pred中的数据
5.UMAP可视化
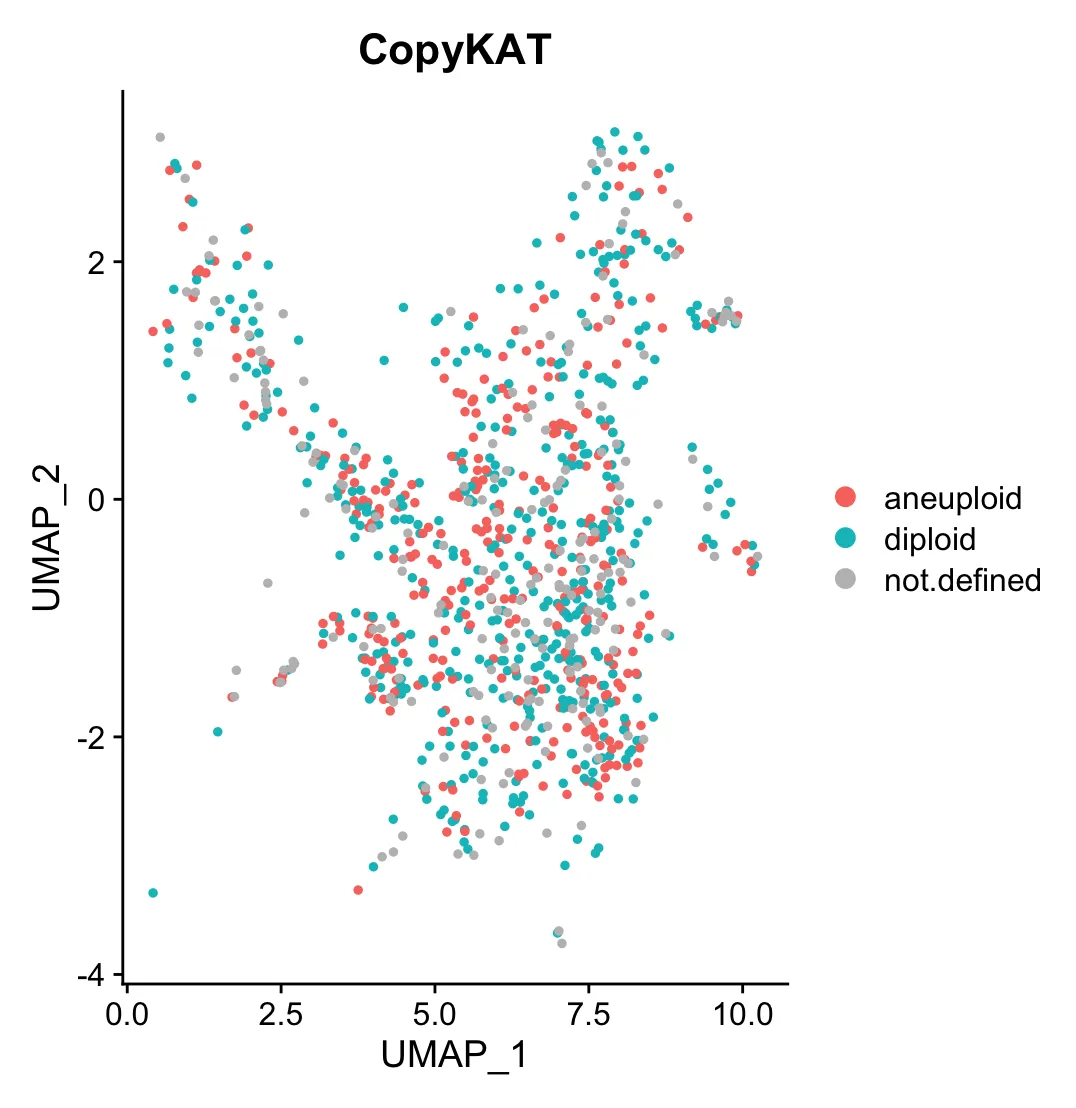
# 把结果添加到Seurat对象的meta.data中
scRNA$CopyKAT = copykat.test$prediction$copykat.pred
DimPlot(scRNA, group.by = "CopyKAT",reduction = 'umap') +
scale_color_manual(values = c("#F8766D",'#02BFC4', "gray"))
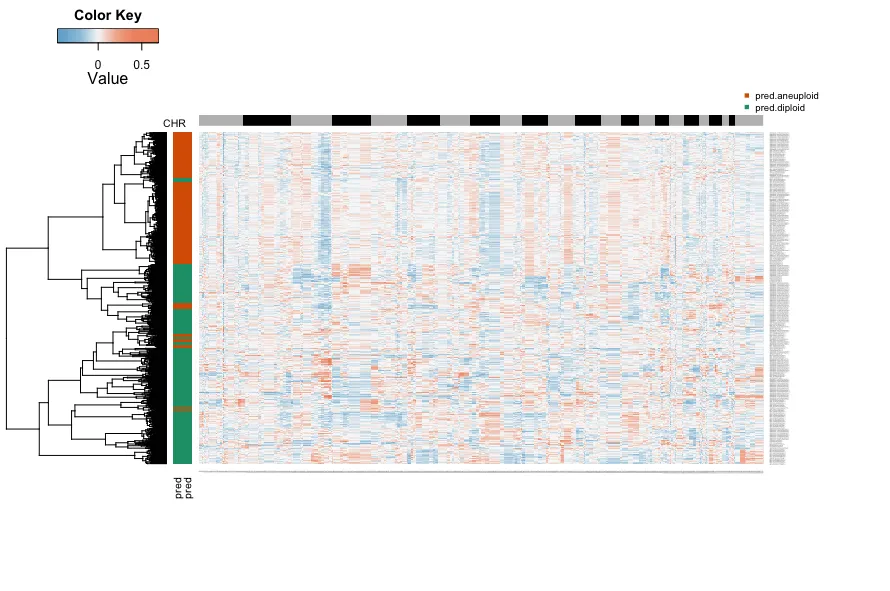
6.热图可视化
开发者给的代码不用改,直接用即可
my_palette <- colorRampPalette(rev(RColorBrewer::brewer.pal(n = 3, name = "RdBu")))(n = 999)
# 染色体编号转化为奇偶性(1和2),用于区分不同的染色体。
chr <- as.numeric(CNA.test$chrom) %% 2+1
# 创建一个黑色到灰色的颜色渐变,用于染色体颜色标注。
rbPal1 <- colorRampPalette(c('black','grey'))
# 为染色体创建一个颜色向量,黑色和灰色表示不同的染色体。
CHR <- rbPal1(2)[as.numeric(chr)]
# 扩展CHR,生成一个二列矩阵,作为热图的ColSideColors参数,用于在热图的列侧边添加染色体颜色标注。
chr1 <- cbind(CHR,CHR)
# 使用Dark2调色板创建一个颜色渐变,用于预测类别的颜色标注
rbPal5 <- colorRampPalette(RColorBrewer::brewer.pal(n = 8, name = "Dark2")[2:1])
# 从pred.test中获取拷贝数预测结果
com.preN <- pred.test$copykat.pred
# 将预测结果转化为颜色(对应于不同的预测类别)。
pred <- rbPal5(2)[as.numeric(factor(com.preN))]
cells <- rbind(pred,pred)
col_breaks = c(seq(-1,-0.4,length=50),seq(-0.4,-0.2,length=150),
seq(-0.2,0.2,length=600),seq(0.2,0.4,length=150),seq(0.4, 1,length=50))
heatmap.3(t(CNA.test[,4:ncol(CNA.test)]),dendrogram="r",
distfun = function(x) parallelDist::parDist(x,threads =4, method = "euclidean"),
hclustfun = function(x) hclust(x, method="ward.D2"),
ColSideColors=chr1,RowSideColors=cells,Colv=NA, Rowv=TRUE,
notecol="black",col=my_palette,breaks=col_breaks, key=TRUE,
keysize=1, density.info="none", trace="none",
cexRow=0.1,cexCol=0.1,cex.main=1,cex.lab=0.1,
symm=F,symkey=F,symbreaks=T,cex=1, cex.main=4, margins=c(10,10))
legend("topright",
paste("pred.",names(table(com.preN)),sep=""),
pch=15,
col=RColorBrewer::brewer.pal(n = 8, name = "Dark2")[2:1],
cex=0.6, bty="n"
)
这里可以看到非整倍体和二倍体的聚类结果,笔者的数据并没有那么明显。使用者在自行分析的时候接下来就可以提起非整倍体细胞进行分析,关于这里的非定义细胞的处理,笔者也没有找到明确处理方式,但不建议纳入分析。
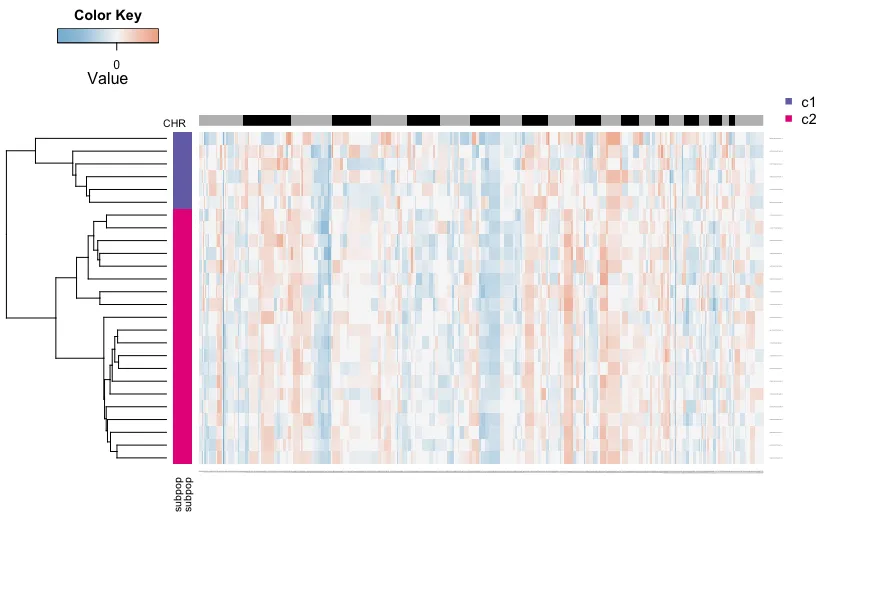
7.定义非整倍体肿瘤细胞的亚群
开发者这里提取了肿瘤的非整倍体细胞
然后用hc.umap <- cutree(hcc,2)划分成两群细胞,使用者可以自行划分~
但一般不用这种方法去划分肿瘤亚群hhh,可以作为辅助验证使用
tumor.cells <- pred.test$cell.names[which(pred.test$copykat.pred=="aneuploid")]
tumor.mat <- CNA.test[, which(colnames(CNA.test) %in% tumor.cells)]
hcc <- hclust(parallelDist::parDist(t(tumor.mat),threads =4, method = "euclidean"),
method = "ward.D2")
hc.umap <- cutree(hcc,2)
rbPal6 <- colorRampPalette(RColorBrewer::brewer.pal(n = 8, name = "Dark2")[3:4])
subpop <- rbPal6(2)[as.numeric(factor(hc.umap))]
cells <- rbind(subpop,subpop)
heatmap.3(t(tumor.mat),dendrogram="r",
distfun = function(x) parallelDist::parDist(x,threads =4, method = "euclidean"),
hclustfun = function(x) hclust(x, method="ward.D2"),
ColSideColors=chr1,RowSideColors=cells,Colv=NA, Rowv=TRUE,
notecol="black",col=my_palette,breaks=col_breaks, key=TRUE,
keysize=1, density.info="none", trace="none",
cexRow=0.1,cexCol=0.1,cex.main=1,cex.lab=0.1,
symm=F,symkey=F,symbreaks=T,cex=1, cex.main=4, margins=c(10,10))
legend("topright", c("c1","c2"),
pch=15,
col=RColorBrewer::brewer.pal(n = 8, name = "Dark2")[3:4],
cex=0.9, bty='n')
开发者不建议使用该工具去预测CNA较少的儿童肿瘤和液体肿瘤中的肿瘤细胞和正常细胞。为解决此问题,CopyKAT提供了两种方法来避免直接无结果的情况并获得一定的输出:1)输入来自同一数据集中已知正常细胞的细胞名称向量;2)通过T细胞。
参考资料:
-
Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat Biotechnol. 2021 May;39(5):599-608.
-
github: https://github.com/navinlabcode/copykat
-
单细胞天地:https://mp.weixin.qq.com/s/7Qz_LSW_cYFR2BL4pS5_NA
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -
原文地址:https://blog.csdn.net/zfyyzhys/article/details/142993875
免责声明:本站文章内容转载自网络资源,如本站内容侵犯了原著者的合法权益,可联系本站删除。更多内容请关注自学内容网(zxcms.com)!