【一只羊做生信】bulk普通转录组上游分析一文搞定
转录组的基本概念
什么是转录组?
转录组是指特定组织或细胞在某一阶段或功能下转录出来的所有RNA的总和,主要包括mRNA和lncRNA
转录组的测定方法?
-
基因芯片(Microarray):
-
基因芯片是一种高通量的技术,可以同时检测成千上万个基因的表达水平。
-
优点:高通量、可以同时检测多个基因、适用于大规模研究。
-
缺点:受限于设计的探针、不能检测未知基因、动态范围较窄。
-
SAGE(Serial Analysis of Gene Expression):
-
SAGE是一种基于串联分析的基因表达技术,可以定量测定基因的表达水平。它在早期被广泛应用,但后来被RNA-seq等新技术所取代。
-
优点:能够定量测定基因表达水平、适用于低丰度基因。
-
缺点:需要大量测序、数据分析复杂、不适用于高通量测序。
-
RNA-seq(RNA sequencing):
-
RNA-seq是一种基于高通量测序技术的方法,可以直接测定RNA的序列和表达水平。它已经成为研究基因表达和转录组学的主流技术。
-
优点:高分辨率、能够检测新基因、广泛应用于转录组学研究。
测序技术及其发展?
-
Sanger测序(第一代测序):
-
发展历程: Sanger测序是由Frederick Sanger在1970年代开发的,是第一种被广泛应用的DNA测序方法。它采用二进制终止方法,使用双脱氧核苷酸(ddNTPs)。
-
优点: 准确可靠,在许多年内被广泛使用。
-
缺点: 吞吐量有限,对于大规模测序项目来说需要耗费大量人力和时间。
-
下一代测序(NGS)- 第二代测序:
-
发展历程: 下一代测序技术,如Illumina测序,于2000年代中期出现,彻底改变了基因组学领域。这些技术提供了高通量测序能力,使DNA和RNA的测序更快速、更具成本效益。
-
优点: 高吞吐量,每碱基成本较低,比Sanger测序具有更快的周转时间。
-
缺点: 短读长,可能存在测序错误,数据分析中存在生物信息学挑战。
-
第三代测序:
-
发展历程: 第三代测序技术,包括PacBio和Oxford Nanopore测序,代表了测序技术的最新进展。这些技术提供了长读长和实时测序能力。
-
优点: 长读长,能够测序基因组的复杂区域,实时测序。
-
缺点: 与NGS技术相比存在更高的错误率,每碱基成本更高,由于错误率较高,数据分析存在挑战。
测序技术研究目标的多样性?
-
基因组学研究: 对不同生物种类的基因组进行测序,揭示基因组结构、功能和进化过程。
-
转录组学研究: 通过RNA测序技术,可以研究基因的表达水平、剪接变体和转录调控网络,从而揭示基因的转录调控机制。
-
表观基因组学研究: 用于研究表观遗传学,包括DNA甲基化和组蛋白修饰等,揭示这些表观遗传标记在基因表达和细胞分化中的作用。
-
微生物组研究: 研究微生物的多样性、功能和在环境和人类健康中的作用。
-
进化生物学研究: 用于研究物种的遗传变异、进化历史和适应性演化,揭示生物多样性的起源和演化过程。
-
生态学研究: 测序技术在生态学研究中也有重要应用,例如分析环境样品中的微生物群落结构、食物网关系等。
-
疾病研究:外显子测序是一种针对基因组中编码蛋白质的外显子区域的测序方法,用于寻找致病突变、研究基因变异与疾病关联等。
二代测序面临的挑战?和发展前景
-
数据处理和存储: 数据量巨大,对数据处理和存储能力提出了挑战,需要强大的计算资源和高效的数据处理算法。
-
错误率: 存在测序错误率,尤其是在重复序列或GC-rich区域容易出现错误,这对数据准确性提出了要求。
-
长读长需求: 对于一些应用,如组装复杂基因组、检测结构变异等,需要更长的读长来解决重复序列和重组事件等问题。
-
成本: 尽管二代测序已经大幅降低了测序成本,但仍然存在一定的成本压力,尤其是在大规模测序项目中。
尽管二代测序技术面临一些挑战,但在单细胞测序等方面仍有着广阔的前景和应用潜力。单细胞测序本公众号已经更新了很多干货内容及优质课程,并且还在继续更新中。相比单细胞测序,普通转录组的分析就要简单许多。本文以水稻普通转录组分析为例,带来其全部的上游分析部分。
环境搭建
点击这里查看环境搭建的基础内容:
开学第一课——从零开始搭建生信分析环境
Mac下生信分析环境搭建
RNA-seq的分析流程通常是这样的:
-
实验设计,提取RNA及文库构建: 确定研究目的、样本类型和处理条件,设计实验方案,包括RNA提取、文库构建和测序平台选择等。从样本中提取总RNA,将RNA反转录为cDNA,然后进行文库构建。
-
测序: 对构建好的RNA文库进行高通量测序,通常使用Illumina等平台进行短读长度测序,生成原始测序数据(raw_data)。在这里要注意,如果使用公共数据进行分析,从NCBI等数据库下载的原始数据很多为SRA格式,需要转换成fastq文件。常用工具为SRA Toolkit,这时还需要对raw_data进行质控。如果是自己测序,下机后得到了fastq的raw_data,通常测序公司在将数据返还给客户之前已经做了“clean”处理,即得到clean_data。本文将跳过质控部分,直接做mapping。
-
序列比对: 将质控后的reads比对到参考基因组或转录组上,利用比对工具(如STAR、HISAT2)进行reads的比对和定位。
-
表达量计数: 根据比对结果,使用计数工具(如featureCounts、HTSeq)计算每个基因的表达量,通常以FPKM(fragments per kilobase of transcript per million mapped reads)或TPM(transcripts per million)为单位。
-
差异表达分析: 使用统计学方法(如DESeq2、edgeR)对不同条件下的基因表达量进行比较,识别差异表达的基因,并进行统计显著性分析。
-
功能注释和富集分析: 对差异表达基因进行功能注释,包括基因本体(Gene Ontology)和通路富集分析,了解不同基因集的生物学功能和通路。
-
可视化和解释: 结果可视化,如热图、火山图等,帮助解释实验结果,并进一步挖掘基因表达的生物学意义。
已经分享过的内容:
关于PC更新R和clusterprofiler包
富集分析踩坑记录|更新clusterprofiler包
水稻进行富集分析
部分可视化方法可在R语言科研作图实例合集中查看。
mamba create -n rna_seq python=2
mamba activate rna_seq
mamba install -y sra-tools fastqc multiqc hisat2 samtools subread gffread
上游分析全流程
本文使用的流程是这样的: fastqc+multiqc(查看质控报告) -> Hisat2 + samtools(比对及转换) -> featureCounts(计数)
创建目录
mkdir ~/2024/rna_seq
cd rna_seq/
mkdir {ref,data,fastqc,align,count}
fastqc+multiqc
fastqc的使用
fastqc *gz -o result/
-q:安静运行,运行过程当中不会生成报告,在结束时将报告生成一个文件
-t参数指定线程数
*gz为所有包含gz的文件
-o 参数输出结果
cd ../fastqc/
fastqc -t 6 -O ./ ../data//bulk_C*/*/*.gz
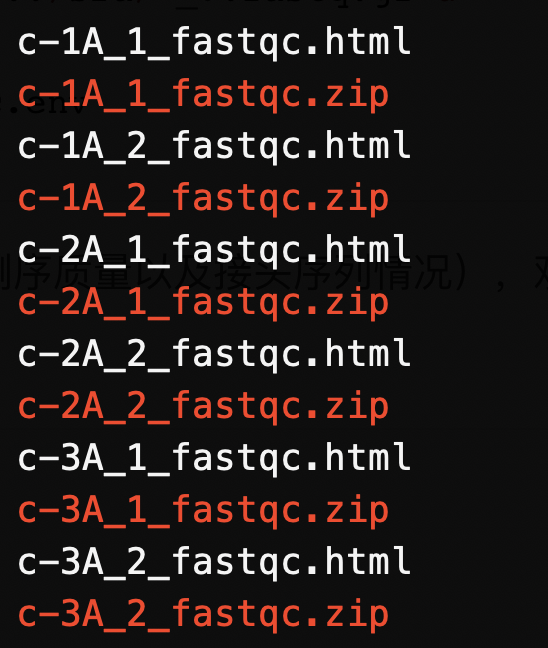
阅读质控报告(重点关注一下测序质量以及接头序列情况),观察是否需要进行截断以及切除接头序列。可以单独点击打开,也可以使用multiqc汇总质控报告
multiqc ./*.zip
Hisat2 + samtools
在进行mapping前首先需要构建index,研究物种的基因组文件下载看这里:
基因组文件的获取 首先需要使用hisat2软件自带的py脚本提取exon和splice的位点
hisat2_extract_exons.py ~/2023/03.ref/Oryza_sativa_IRGSP/Oryza_sativa.IRGSP-1.0.56.gtf > genome.exon
hisat2_extract_splice_sites.py ~/2023/03.ref/Oryza_sativa_IRGSP/Oryza_sativa.IRGSP-1.0.56.gtf > genome.ss
进行index的构建,hisat2-build的完整说明
Usage: hisat2-build [options]* <reference_in> <ht2_index_base>
reference_in comma-separated list of files with ref sequences
hisat2_index_base write ht2 data to files with this dir/basename
Options:
-c reference sequences given on cmd line (as
<reference_in>)
--large-index force generated index to be 'large', even if ref
has fewer than 4 billion nucleotides
-a/--noauto disable automatic -p/--bmax/--dcv memory-fitting
**-p <int> number of threads**
--bmax <int> max bucket sz for blockwise suffix-array builder
--bmaxdivn <int> max bucket sz as divisor of ref len (default: 4)
--dcv <int> diff-cover period for blockwise (default: 1024)
--nodc disable diff-cover (algorithm becomes quadratic)
-r/--noref don't build .3/.4.ht2 (packed reference) portion
-3/--justref just build .3/.4.ht2 (packed reference) portion
-o/--offrate <int> SA is sampled every 2^offRate BWT chars (default: 5)
-t/--ftabchars <int> # of chars consumed in initial lookup (default: 10)
--localoffrate <int> SA (local) is sampled every 2^offRate BWT chars (default: 3)
--localftabchars <int> # of chars consumed in initial lookup in a local index (default: 6)
--snp <path> SNP file name
--haplotype <path> haplotype file name
--ss <path> Splice site file name
--exon <path> Exon file name
--repeat-ref <path> Repeat reference file name
--repeat-info <path> Repeat information file name
--repeat-snp <path> Repeat snp file name
--repeat-haplotype <path> Repeat haplotype file name
--seed <int> seed for random number generator
-q/--quiet disable verbose output (for debugging)
-h/--help print detailed description of tool and its options
--usage print this usage message
--version print version information and quit
hisat2-build -p 20 ~/03.ref/Oryza_sativa_IRGSP/Oryza_sativa.IRGSP-1.0.dna.toplevel.fa --ss genome.ss --exon genome.exon Oryza_sativa_IRGSP
hisat2命令的完整说明
Usage:
hisat2 [options]* -x <ht2-idx> {-1 <m1> -2 <m2> | -U <r>} [-S <sam>]
<ht2-idx> Index filename prefix (minus trailing .X.ht2).
<m1> Files with #1 mates, paired with files in <m2>.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<m2> Files with #2 mates, paired with files in <m1>.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<r> Files with unpaired reads.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<sam> File for SAM output (default: stdout)
<m1>, <m2>, <r> can be comma-separated lists (no whitespace) and can be
specified many times. E.g. '-U file1.fq,file2.fq -U file3.fq'.
Options (defaults in parentheses):
Input:
-q query input files are FASTQ .fq/.fastq (default)
--qseq query input files are in Illumina's qseq format
-f query input files are (multi-)FASTA .fa/.mfa
-r query input files are raw one-sequence-per-line
-c <m1>, <m2>, <r> are sequences themselves, not files
-s/--skip <int> skip the first <int> reads/pairs in the input (none)
-u/--upto <int> stop after first <int> reads/pairs (no limit)
-5/--trim5 <int> trim <int> bases from 5'/left end of reads (0)
-3/--trim3 <int> trim <int> bases from 3'/right end of reads (0)
--phred33 qualities are Phred+33 (default)
--phred64 qualities are Phred+64
--int-quals qualities encoded as space-delimited integers
Presets: Same as:
--fast --no-repeat-index
--sensitive --bowtie2-dp 1 -k 30 --score-min L,0,-0.5
--very-sensitive --bowtie2-dp 2 -k 50 --score-min L,0,-1
Alignment:
--bowtie2-dp <int> use Bowtie2's dynamic programming alignment algorithm (0) - 0: no dynamic programming, 1: conditional dynamic programming, and 2: unconditional dynamic programming (slowest)
--n-ceil <func> func for max # non-A/C/G/Ts permitted in aln (L,0,0.15)
--ignore-quals treat all quality values as 30 on Phred scale (off)
--nofw do not align forward (original) version of read (off)
--norc do not align reverse-complement version of read (off)
--no-repeat-index do not use repeat index
Spliced Alignment:
--pen-cansplice <int> penalty for a canonical splice site (0)
--pen-noncansplice <int> penalty for a non-canonical splice site (12)
--pen-canintronlen <func> penalty for long introns (G,-8,1) with canonical splice sites
--pen-noncanintronlen <func> penalty for long introns (G,-8,1) with noncanonical splice sites
--min-intronlen <int> minimum intron length (20)
--max-intronlen <int> maximum intron length (500000)
--known-splicesite-infile <path> provide a list of known splice sites
--novel-splicesite-outfile <path> report a list of splice sites
--novel-splicesite-infile <path> provide a list of novel splice sites
--no-temp-splicesite disable the use of splice sites found
--no-spliced-alignment disable spliced alignment
--rna-strandness <string> specify strand-specific information (unstranded)
--tmo reports only those alignments within known transcriptome
--dta reports alignments tailored for transcript assemblers
--dta-cufflinks reports alignments tailored specifically for cufflinks
--avoid-pseudogene tries to avoid aligning reads to pseudogenes (experimental option)
--no-templatelen-adjustment disables template length adjustment for RNA-seq reads
Scoring:
--mp <int>,<int> max and min penalties for mismatch; lower qual = lower penalty <6,2>
--sp <int>,<int> max and min penalties for soft-clipping; lower qual = lower penalty <2,1>
--no-softclip no soft-clipping
--np <int> penalty for non-A/C/G/Ts in read/ref (1)
--rdg <int>,<int> read gap open, extend penalties (5,3)
--rfg <int>,<int> reference gap open, extend penalties (5,3)
--score-min <func> min acceptable alignment score w/r/t read length
(L,0.0,-0.2)
Reporting:
-k <int> It searches for at most <int> distinct, primary alignments for each read. Primary alignments mean
alignments whose alignment score is equal to or higher than any other alignments. The search terminates
when it cannot find more distinct valid alignments, or when it finds <int>, whichever happens first.
The alignment score for a paired-end alignment equals the sum of the alignment scores of
the individual mates. Each reported read or pair alignment beyond the first has the SAM ‘secondary’ bit
(which equals 256) set in its FLAGS field. For reads that have more than <int> distinct,
valid alignments, hisat2 does not guarantee that the <int> alignments reported are the best possible
in terms of alignment score. Default: 5 (linear index) or 10 (graph index).
Note: HISAT2 is not designed with large values for -k in mind, and when aligning reads to long,
repetitive genomes, large -k could make alignment much slower.
--max-seeds <int> HISAT2, like other aligners, uses seed-and-extend approaches. HISAT2 tries to extend seeds to
full-length alignments. In HISAT2, --max-seeds is used to control the maximum number of seeds that
will be extended. For DNA-read alignment (--no-spliced-alignment), HISAT2 extends up to these many seeds
and skips the rest of the seeds. For RNA-read alignment, HISAT2 skips extending seeds and reports
no alignments if the number of seeds is larger than the number specified with the option,
to be compatible with previous versions of HISAT2. Large values for --max-seeds may improve alignment
sensitivity, but HISAT2 is not designed with large values for --max-seeds in mind, and when aligning
reads to long, repetitive genomes, large --max-seeds could make alignment much slower.
The default value is the maximum of 5 and the value that comes with -k times 2.
-a/--all HISAT2 reports all alignments it can find. Using the option is equivalent to using both --max-seeds
and -k with the maximum value that a 64-bit signed integer can represent (9,223,372,036,854,775,807).
--repeat report alignments to repeat sequences directly
Paired-end:
-I/--minins <int> minimum fragment length (0), only valid with --no-spliced-alignment
-X/--maxins <int> maximum fragment length (500), only valid with --no-spliced-alignment
--fr/--rf/--ff -1, -2 mates align fw/rev, rev/fw, fw/fw (--fr)
--no-mixed suppress unpaired alignments for paired reads
--no-discordant suppress discordant alignments for paired reads
Output:
-t/--time print wall-clock time taken by search phases
--un <path> write unpaired reads that didn't align to <path>
--al <path> write unpaired reads that aligned at least once to <path>
--un-conc <path> write pairs that didn't align concordantly to <path>
--al-conc <path> write pairs that aligned concordantly at least once to <path>
(Note: for --un, --al, --un-conc, or --al-conc, add '-gz' to the option name, e.g.
--un-gz <path>, to gzip compress output, or add '-bz2' to bzip2 compress output.)
--summary-file <path> print alignment summary to this file.
--new-summary print alignment summary in a new style, which is more machine-friendly.
--quiet print nothing to stderr except serious errors
--met-file <path> send metrics to file at <path> (off)
--met-stderr send metrics to stderr (off)
--met <int> report internal counters & metrics every <int> secs (1)
--no-head suppress header lines, i.e. lines starting with @
--no-sq suppress @SQ header lines
--rg-id <text> set read group id, reflected in @RG line and RG:Z: opt field
--rg <text> add <text> ("lab:value") to @RG line of SAM header.
Note: @RG line only printed when --rg-id is set.
--omit-sec-seq put '*' in SEQ and QUAL fields for secondary alignments.
Performance:
-o/--offrate <int> override offrate of index; must be >= index's offrate
-p/--threads <int> number of alignment threads to launch (1)
--reorder force SAM output order to match order of input reads
--mm use memory-mapped I/O for index; many 'hisat2's can share
Other:
--qc-filter filter out reads that are bad according to QSEQ filter
--seed <int> seed for random number generator (0)
--non-deterministic seed rand. gen. arbitrarily instead of using read attributes
--remove-chrname remove 'chr' from reference names in alignment
--add-chrname add 'chr' to reference names in alignment
--version print version information and quit
-h/--help print this usage message
示例代码
for i in `ls ../data/bulk_C*/*/*_1*.gz | cut -d "/" -f 4`
do
hisat2 -p 20 -x ../ref/Oryza_sativa_IRGSP -1 ../data/bulk_C*/${i}/${i}_1.fq.gz -2 ../data/bulk_C*/${i}/${i}_2.fq.gz -S ./${i}.sam
done
在这里可以写一个shell脚本然后投递到后台慢慢运行。
SAM转BAM
samtools的使用方法:
Usage: samtools sort [options...] [in.bam]
Options:
-l INT Set compression level, from 0 (uncompressed) to 9 (best)
-m INT Set maximum memory per thread; suffix K/M/G recognized [768M]
-n Sort by read name
-t TAG Sort by value of TAG. Uses position as secondary index (or read name if -n is set)
-o FILE Write final output to FILE rather than standard output
-T PREFIX Write temporary files to PREFIX.nnnn.bam
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
**-O, --output-fmt FORMAT[,OPT[=VAL]]...**
Specify output format (SAM, BAM, CRAM)
--output-fmt-option OPT[=VAL]
Specify a single output file format option in the form
of OPTION or OPTION=VALUE
--reference FILE
Reference sequence FASTA FILE [null]
**-@, --threads INT**
Number of additional threads to use [0]
示例代码:
ls *.sam|while read i
do
samtools sort -O bam -@ 20 -o ${i%%.*}.bam ${i}
done
rm *.sam#删除掉所有的sam文件,因为sam文件占的内存太大
批量进行bam索引的构建
ls *.bam|xargs -i samtools index {}
featureCounts
featureCounts的使用参数
\## Mandatory arguments:
-a <string> Name of an annotation file. GTF/GFF format by default. See
-F option for more format information. Inbuilt annotations
(SAF format) is available in 'annotation' directory of the
package. Gzipped file is also accepted.
-o <string> Name of output file including read counts. A separate file
including summary statistics of counting results is also
included in the output ('<string>.summary'). Both files
are in tab delimited format.
input_file1 [input_file2] ... A list of SAM or BAM format files. They can be
either name or location sorted. If no files provided,
<stdin> input is expected. Location-sorted paired-end reads
are automatically sorted by read names.
\## Optional arguments:
\# Annotation
-F <string> Specify format of the provided annotation file. Acceptable
formats include 'GTF' (or compatible GFF format) and
'SAF'. 'GTF' by default. For SAF format, please refer to
Users Guide.
**-t <string> Specify feature type(s) in a GTF annotation. If multiple**
**types are provided, they should be separated by ',' with**
**no space in between. 'exon' by default. Rows in the**
**annotation with a matched feature will be extracted and**
**used for read mapping.**
-g <string> Specify attribute type in GTF annotation. 'gene_id' by
default. Meta-features used for read counting will be
extracted from annotation using the provided value.
--extraAttributes Extract extra attribute types from the provided GTF
annotation and include them in the counting output. These
attribute types will not be used to group features. If
more than one attribute type is provided they should be
separated by comma.
-A <string> Provide a chromosome name alias file to match chr names in
annotation with those in the reads. This should be a two-
column comma-delimited text file. Its first column should
include chr names in the annotation and its second column
should include chr names in the reads. Chr names are case
sensitive. No column header should be included in the
file.
\# Level of summarization
-f Perform read counting at feature level (eg. counting
reads for exons rather than genes).
\# Overlap between reads and features
-O Assign reads to all their overlapping meta-features (or
features if -f is specified).
--minOverlap <int> Minimum number of overlapping bases in a read that is
required for read assignment. 1 by default. Number of
overlapping bases is counted from both reads if paired
end. If a negative value is provided, then a gap of up
to specified size will be allowed between read and the
feature that the read is assigned to.
--fracOverlap <float> Minimum fraction of overlapping bases in a read that is
required for read assignment. Value should be within range
[0,1]. 0 by default. Number of overlapping bases is
counted from both reads if paired end. Both this option
and '--minOverlap' option need to be satisfied for read
assignment.
--fracOverlapFeature <float> Minimum fraction of overlapping bases in a
feature that is required for read assignment. Value
should be within range [0,1]. 0 by default.
--largestOverlap Assign reads to a meta-feature/feature that has the
largest number of overlapping bases.
--nonOverlap <int> Maximum number of non-overlapping bases in a read (or a
read pair) that is allowed when being assigned to a
feature. No limit is set by default.
--nonOverlapFeature <int> Maximum number of non-overlapping bases in a feature
that is allowed in read assignment. No limit is set by
default.
--readExtension5 <int> Reads are extended upstream by <int> bases from their
5' end.
--readExtension3 <int> Reads are extended upstream by <int> bases from their
3' end.
--read2pos <5:3> Reduce reads to their 5' most base or 3' most base. Read
counting is then performed based on the single base the
read is reduced to.
\# Multi-mapping reads
-M Multi-mapping reads will also be counted. For a multi-
mapping read, all its reported alignments will be
counted. The 'NH' tag in BAM/SAM input is used to detect
multi-mapping reads.
\# Fractional counting
--fraction Assign fractional counts to features. This option must
be used together with '-M' or '-O' or both. When '-M' is
specified, each reported alignment from a multi-mapping
read (identified via 'NH' tag) will carry a fractional
count of 1/x, instead of 1 (one), where x is the total
number of alignments reported for the same read. When '-O'
is specified, each overlapping feature will receive a
fractional count of 1/y, where y is the total number of
features overlapping with the read. When both '-M' and
'-O' are specified, each alignment will carry a fractional
count of 1/(x*y).
\# Read filtering
-Q <int> The minimum mapping quality score a read must satisfy in
order to be counted. For paired-end reads, at least one
end should satisfy this criteria. 0 by default.
--splitOnly Count split alignments only (ie. alignments with CIGAR
string containing 'N'). An example of split alignments is
exon-spanning reads in RNA-seq data.
--nonSplitOnly If specified, only non-split alignments (CIGAR strings do
not contain letter 'N') will be counted. All the other
alignments will be ignored.
--primary Count primary alignments only. Primary alignments are
identified using bit 0x100 in SAM/BAM FLAG field.
--ignoreDup Ignore duplicate reads in read counting. Duplicate reads
are identified using bit Ox400 in BAM/SAM FLAG field. The
whole read pair is ignored if one of the reads is a
duplicate read for paired end data.
\# Strandness
-s <int or string> Perform strand-specific read counting. A single integer
value (applied to all input files) or a string of comma-
separated values (applied to each corresponding input
file) should be provided. Possible values include:
0 (unstranded), 1 (stranded) and 2 (reversely stranded).
Default value is 0 (ie. unstranded read counting carried
out for all input files).
\# Exon-exon junctions
-J Count number of reads supporting each exon-exon junction.
Junctions were identified from all the exon-spanning reads
in the input (containing 'N' in CIGAR string). Counting
results are saved to a file named '<output_file>.jcounts'
-G <string> Provide the name of a FASTA-format file that contains the
reference sequences used in read mapping that produced the
provided SAM/BAM files. This optional argument can be used
with '-J' option to improve read counting for junctions.
\# Parameters specific to paired end reads
-p If specified, libraries are assumed to contain paired-end
reads. For any library that contains paired-end reads, the
'countReadPairs' parameter controls if read pairs or reads
should be counted.
--countReadPairs If specified, fragments (or templates) will be counted
instead of reads. This option is only applicable for
paired-end reads. For single-end data, it is ignored.
-B Only count read pairs that have both ends aligned.
-P Check validity of paired-end distance when counting read
pairs. Use -d and -D to set thresholds.
-d <int> Minimum fragment/template length, 50 by default.
-D <int> Maximum fragment/template length, 600 by default.
-C Do not count read pairs that have their two ends mapping
to different chromosomes or mapping to same chromosome
but on different strands.
--donotsort Do not sort reads in BAM/SAM input. Note that reads from
the same pair are required to be located next to each
other in the input.
\# Number of CPU threads
**-T <int> Number of the threads. 1 by default.**
\# Read groups
--byReadGroup Assign reads by read group. "RG" tag is required to be
present in the input BAM/SAM files.
\# Long reads
-L Count long reads such as Nanopore and PacBio reads. Long
read counting can only run in one thread and only reads
(not read-pairs) can be counted. There is no limitation on
the number of 'M' operations allowed in a CIGAR string in
long read counting.
\# Assignment results for each read
-R <format> Output detailed assignment results for each read or read-
pair. Results are saved to a file that is in one of the
following formats: CORE, SAM and BAM. See Users Guide for
more info about these formats.
--Rpath <string> Specify a directory to save the detailed assignment
results. If unspecified, the directory where counting
results are saved is used.
\# Miscellaneous
--tmpDir <string> Directory under which intermediate files are saved (later
removed). By default, intermediate files will be saved to
the directory specified in '-o' argument.
--maxMOp <int> Maximum number of 'M' operations allowed in a CIGAR
string. 10 by default. Both 'X' and '=' are treated as 'M'
and adjacent 'M' operations are merged in the CIGAR
string.
--verbose Output verbose information for debugging, such as un-
matched chromosome/contig names.
-v Output version of the program.
参考代码:
featureCounts -T 20 -p -t exon -g gene_id -a ~/2023/03.ref/Oryza_sativa_IRGSP/Oryza_sativa.IRGSP-1.0.56.gtf -o ../count/expr_matrix.txt ./*.bam
写在文后
以上得到的expr_matrix.txt即为所需要的count值,至此普通转录组上游分析基本结束,代码运行是比较简单但需要分步操作又是有点繁琐的。后续将继续带来普通转录组联合单细胞转录组分析的内容,如果本期文字对您的帮助有帮助,请点个“赞”和“在看”哦~
本篇完。
原文地址:https://blog.csdn.net/jianyangmaodemie/article/details/136711340
免责声明:本站文章内容转载自网络资源,如本站内容侵犯了原著者的合法权益,可联系本站删除。更多内容请关注自学内容网(zxcms.com)!