生信数据分析——绘制差异热图
生信数据分析——绘制差异热图
差异分析的热图(Heat Map)在生物信息学数据分析中,特别是在基因表达差异分析中,是一个非常直观和有用的工具。
本教程将从导入的数据结构开始,一步步带大家在R中绘制好看的热图,最后对热图进行解读,确保读者理解代码的同时学会解读热图里的每个元素所代表的意思。
首先来看一下最终的热图长啥样:
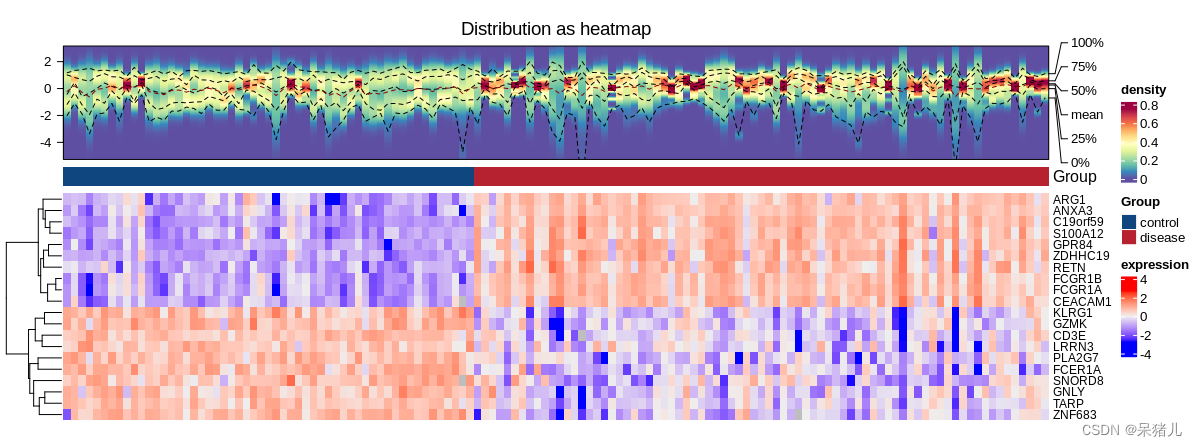
热图的构建基于两个主要的数据:
表达矩阵:行为基因,列为样本的表达矩阵,只要是能反应每个样本不同基因表达水平即可。
分组信息表:与样本对应,要明确知道每个样本属于哪个条件分组。
注:绘制差异热图不需要用到差异分析的结果,但我这里仅展示上下调前10个基因的表达热图,所以用到了差异分析的结果
本项目以GSE73461数据集(芯片数据)展示热图绘制过程
实验分组:疾病组(77例),对照组(55例)
R版本:4.2.2
R包:ComplexHeatmap,tidyverse
废话不多说,代码如下:
设置工作空间:
rm(list = ls()) # 删除工作空间中所有的对象
setwd('/XX/XX/XX') # 设置工作路径
if(!dir.exists('./01_DEGs')){
dir.create('./01_DEGs')
} # 判断该工作路径下是否存在名为01_DEGs的文件夹,如果不存在则创建,如果存在则pass
setwd('./01_DEGs/') # 设置路径到刚才新建的01_DEGs下
加载包:
library(ComplexHeatmap)
library(tidyverse)
导入表达矩阵:train_data
注:芯片数据处理过程参考之前的教程——数据处理(GEO数据库——芯片数据)
train_data <- read.csv('../../00_rawdata/GSE73461/dat.GSE73461.csv', row.names = 1, check.names = F)
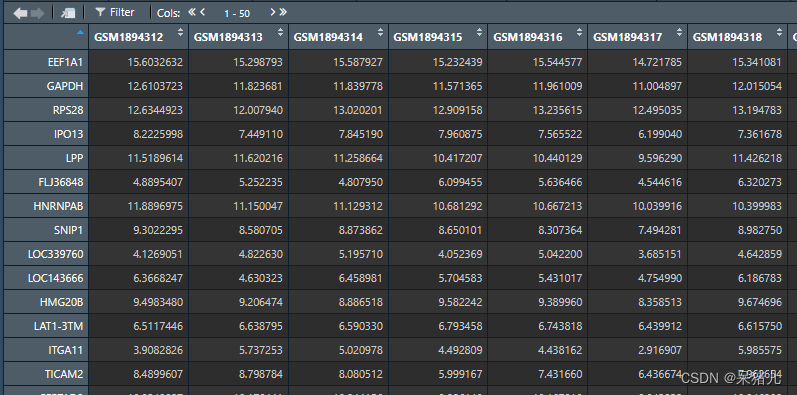
train_data如下图所示,行名是基因,列名是样本名,图中数字代表基因表达量,这样就能获取到每个基因在不同样本中的表达。
导入分组信息表:group,并根据分组信息简单排序
group <- read.csv('../../00_rawdata/GSE73461/group.GSE73461.csv')
group <- group[order(group$group), ] ## 根据分组信息简单排序
table(group$group)
train_data <- train_data[, group$sample] ## 保证表达矩阵与这个分组信息表对应
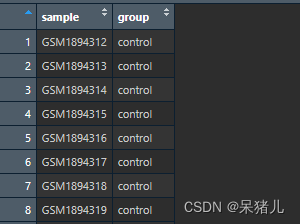
group 如下图所示,第一列是样本ID,第二列是样本对应的分组,这样就能获取到每个样本对应的条件分组。
导入差异分析结果(limma/DESeq2都可以):
DEG <- read.csv('../../01_DEGs/GSE73461/DEG_all.csv'原文地址:https://blog.csdn.net/weixin_49878699/article/details/136178777
免责声明:本站文章内容转载自网络资源,如本站内容侵犯了原著者的合法权益,可联系本站删除。更多内容请关注自学内容网(zxcms.com)!